JGI Seed Corn Funding Project Blog 2022-2023: Sion Bayliss and Daniel Lawson
The future of disease control relies heavily on understanding the evolution and emergence of bacterial strains in real time, a daunting yet crucial task. This project marked a significant step towards this goal, aiming to rapidly unmask potentially problematic ‘hybrids’ caused by the crossing of already established disease lineages and demystify the evolution and adaptation of harmful bacteria. We aim to provide the first steps in an early warning system against potential public health threats posed by new bacterial strains.
Understanding Bacterial Evolution: The Key to Future Disease Outbreaks
Genomic sequencing has become a key part of research programmes worldwide, notably pathogens that affect human and animal health and related strains that exist in the wild. This has led to the deposition of vast numbers of disease genomes in public repositories, a rich resource for the study of evolutionary processes underpinning the emergence of new bacterial lineages. During this project we developed a tool which could be used to search within these large collections and identify potentially problematic strains of disease-causing bacteria.
The focus of the work was on differentiating between whether emerging lineages had predominantly evolved due to hybridisation between of existing lineages or were a product of transfer from novel sources, such as animals or the environment. This differentiation was achieved by scrutinizing the bacterium’s genome sequences, enabling us to identify hybridized DNA sections, a critical step towards comprehending bacterial evolution.
Outcomes: A Rapid Detection System for Dangerous Hybrid Lineages
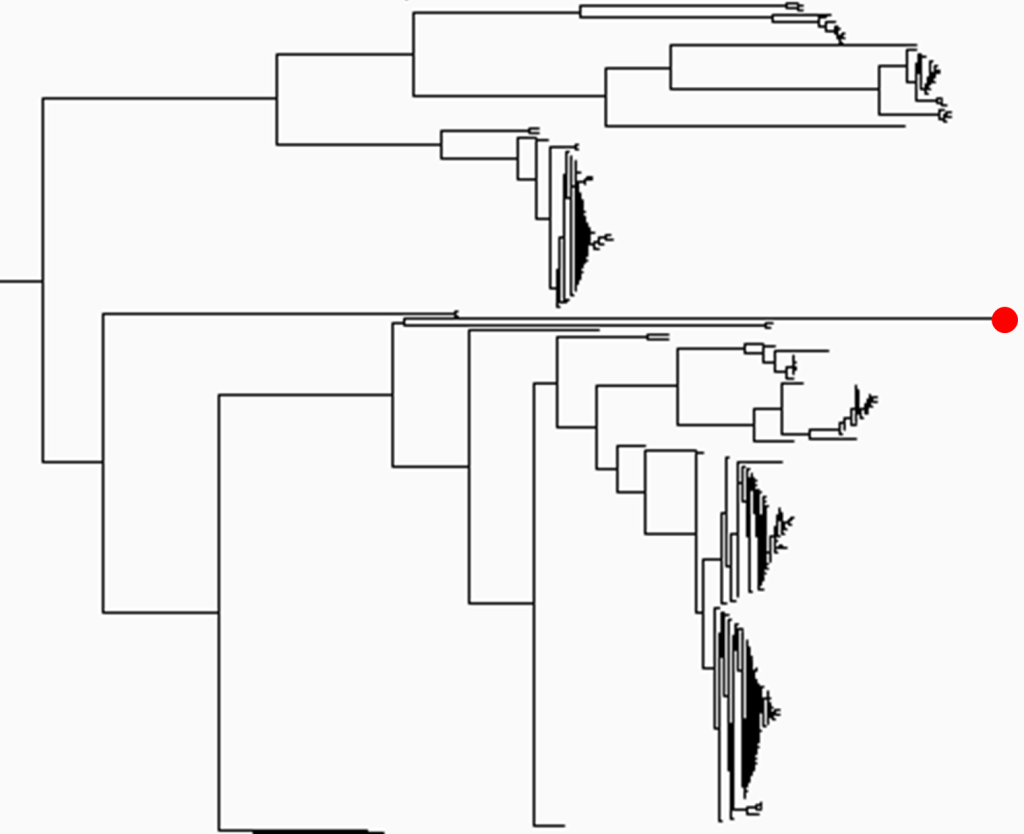
To classify lineages, we clustered sample based upon their ‘family-tree’. By examining these ‘phylogenetic trees’ we are able to identify candidate hybrid lineages present on distinctive long-branches (Figure 1). These ‘long-branch lineages’ could have evolved in various ways – having an increased evolutionary rate, being imported from a previously unknown source, or having recently emerged via hybridisation between two or more parent lineages.
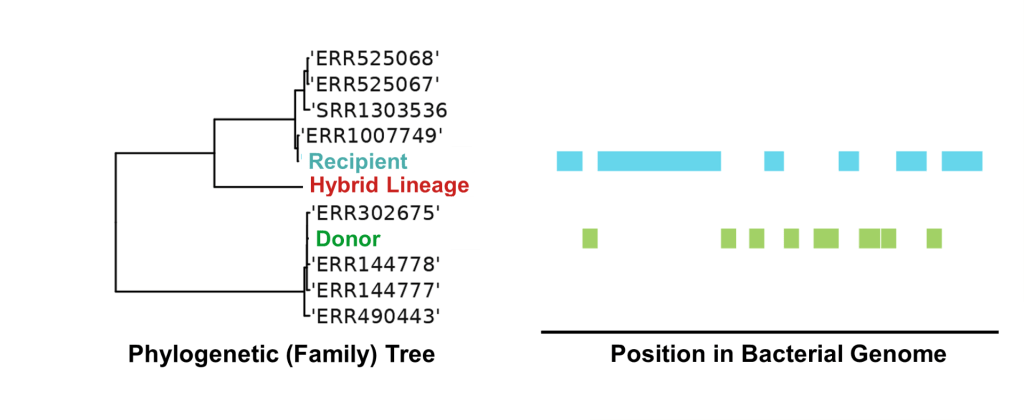
Our groundbreaking achievement lies in the development of a software pipeline capable of rapidly differentiating between these various scenarios. Tested on both simulated and real-world genomic datasets, our software can identify otherwise cryptic ‘hybrid’ lineages (Figure 2). This development lays the foundation for a software tool to ‘flag’ potentially alarming strains being routinely added to large genomic databases which could pose a significant public health threat.
Future Developments
Our ultimate goal is to equip researchers and healthcare professionals with a tool that can provide early warnings for new and potentially dangerous bacterial strains. To this end, our project’s next steps include testing the tool on large and diverse databases of pathogens of public health concern, updating and streamlining the codebase for broader and easier use by other researchers, and development of a web-portal that would allow users to upload their samples for testing against a comprehensive example datasets.
Staying Connected
For more details about our project and future updates, please contact either Sion Bayliss or Daniel Lawson, and feel free to engage with us for any queries or discussions.